
Research
I. Chemistry in Restricted Dimension
.jpg)
.jpg)
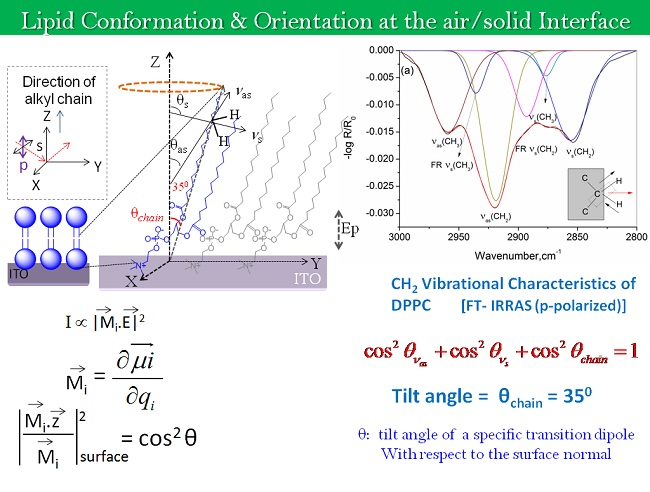
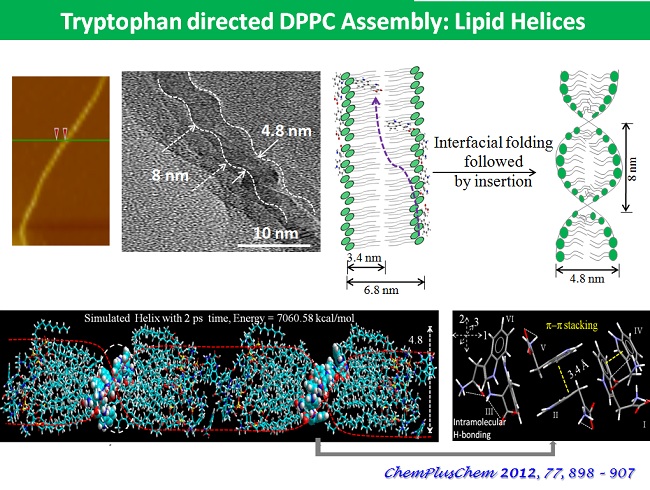
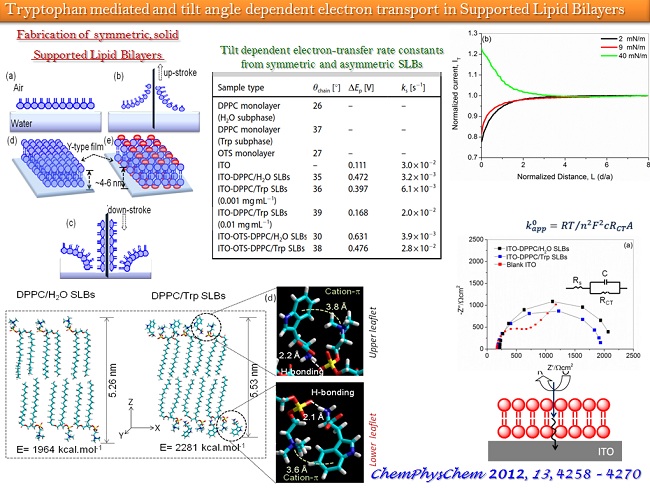
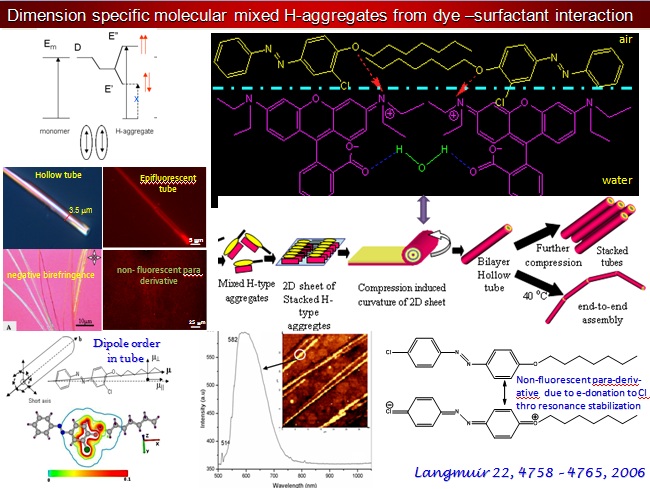
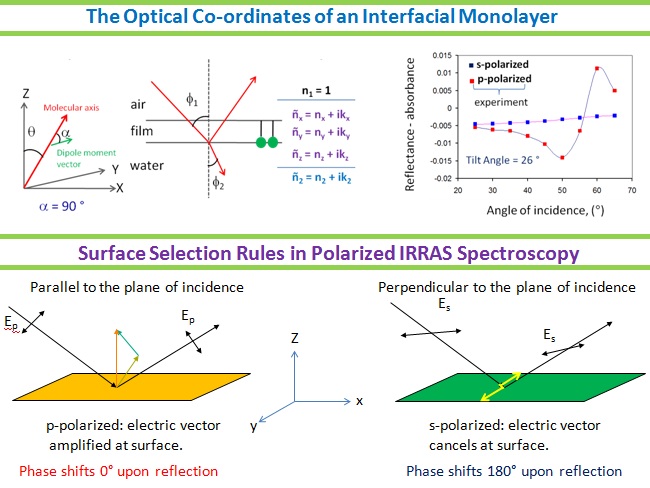
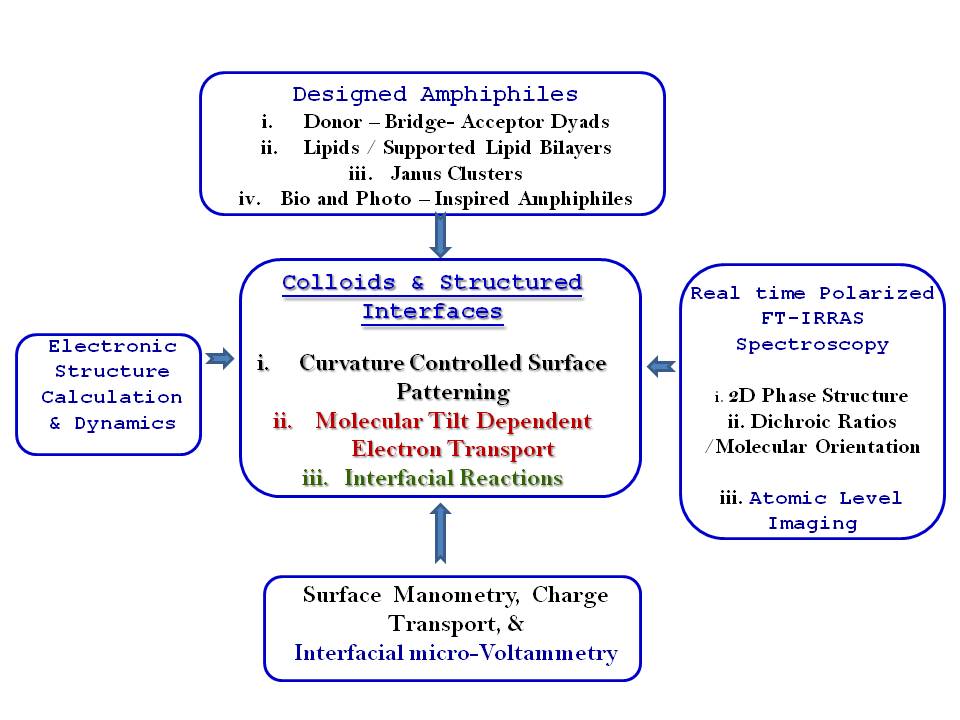
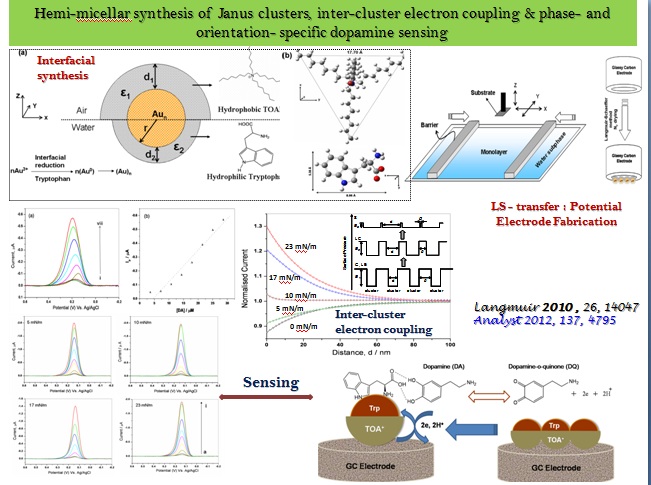
In the ambit of 2D Interfacial Chemistry, we investigate dynamic behavior of monolayer assemblies at liquid/gas and solid/gas interfaces and intend understanding how composition and structure of monolayer assemblies affect the dynamics of lateral molecular diffusion, long range electron tunneling and vectorial electron transport across the anisotropic medium. The goals address issues relevant to the behavior of biological membranes along with reactive interfaces for fabrication of functional electronic materials as sensors and devices. The Langmuir techniques, Scanning Probe Microscopy, 2D electrochemical methods, and high resolution polarized spectroscopy, both in-situ and off -line, are used to study molecular conformations, lateral electron transport in bifunctional Janus clusters, Donor-Bridge-Acceptor dyads and lipid head group – amino acid interactions with model lipid membranes.
Our interest also lies in probing the water liquid-vapor interfacial region with its dynamic properties and understand how they differ from the structure of bulk water. In addition, we want to understand the types of interactions that determine the phase behavior, orientation and the lateral dynamics of bio-mimetic monolayers on the water surface. Our initial efforts on the preparation and evolution of organometallic molecular arrays may lead to materials with promising electro-optic and electronic functions.
II. DFT and semi-empirical ground and excited state electronic structure calculation
ab-initio and semi-empirical ground state electronic structure calculations, incorporating non-covalent interactions (H-bonding, dispersion interactions and the solvent polarity), time dependent density functional calculations with appropriate basis sets, and constrained and unconstrained molecular dynamics simulations have been underway.
Some salient findings:
* mechanistic routes to the exclusively formed 2D soft Ellipsoids from a Benzoic acid amphiphile were established upon H-bonded dimeric structure calculations through potential energy land scapes.
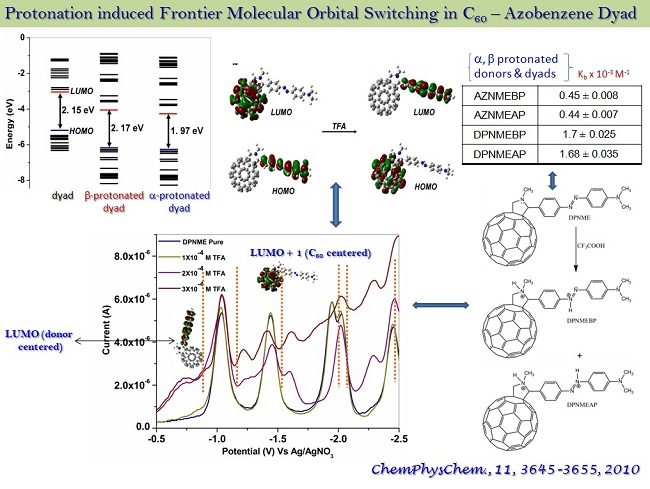
* structure and spectroscopically forbidden transitions in the H- dimeric aggregates of a C60 - Azobenzene dyad were corroborated. Characteristic CT transitions have been a continual theme demanding reform in TD-DFT calculations.
* the role of frontier molecular orbitals in directed electron transport across a C60 based acceptor - bridge - donor dyad was established.
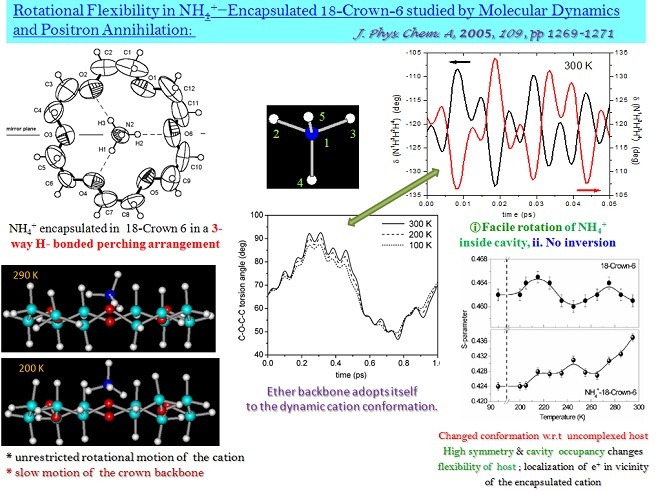
* the computed rotational flexibility of NH4+ ion in NH4+@18-Crown-6 complex was corroborated with parameters obtained from positron lifetime spectroscopy experiments.
III. Solution Phase Aggregate Curvature and Dynamics:
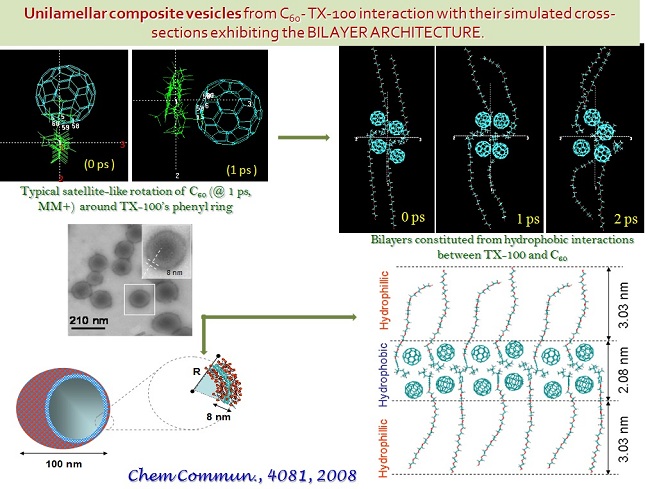
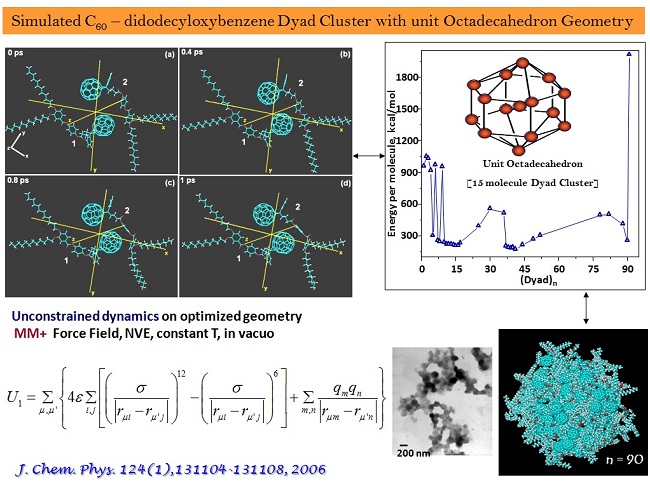
Systematic work leading to understanding the structural needs for the formation of supramolecular containers, such as, persistent micelles, vesicles and assembles of varied aggregate curvature from chiral amphiphiles and surfactants (cationic, anionic, and cationaionic), and hydrophobic - hydrophilic - hydrophobic molecular tectons, satisfying the geometry and thermodynamic requirements have been underway.
Realization of dye-dye aggregate formation in artificially synthesized dyes has been of continued interest in view of new avenues for electronic and photonic materials. Fullerene C60 being a fascinating 3D molecular entity with rich optical and electronic properties, combining it with a photoactive azobenzene chromophore yielded molecular hybrids with novel photochemical, electrochemical and electronic properties. Tunable electronic properties of such a proton responsive C60 dyad exhibiting frontier molecular orbital switching was observed. Our results on the synthesis, molecular modeling and spectroscopy on diverse molecular systems have provided fundamental understandings that may facilitate realization of molecular machines, responding to specific stimuli towards fulfilling specific functions for Nanotechnology.
 Back To Top
Back To Top